
SAMED and SALDA hosted a Regulatory Forum on 5 August 2020 in order to strengthen the industry’s understanding of the vigilance and recall processes for medical technology.
Senior Manager of Medical Devices and Radiation Control at SAHPRA, Ms Andrea Julsing Keyter, presented at the Forum. She said that while not all adverse events need to be reported, it appears that there is under-reporting happening as SAHPRA has received too few reports given that it has 1 800 licenses spanning thousands of products. “We should be getting more reports.”
This could be because licence holders were unsure on what to report or due to fear of repercussions. Ms Julsing Keyter indicated that not every report results in a recall and urged licence holders not to be fearful of over-reporting. Rather suppliers should view the reporting as a means for improving product quality, safety and effectiveness.
Ms Julsing Keyter explained the basic reporting criteria and exemption rules. Specific points to note:
- Recurring adverse events are not exempt as these may indicate trends that require corrective actions.
- When exemptions apply, it is essential that the licence holders still document the incident within their quality management processes in order to monitor and track trends.
- Industry need to follow the regulations on reporting timelines:
- If the event led/could lead to serious threat to public health: reporting to be done 48 hours after the person becomes aware of the event or occurrence.
- If the event led to a death or a serious deterioration in the state of health: reporting within 10 calendar days after the person becomes aware of the event.
- If an event which took place could potentially cause death or a serious deterioration in the state of health: reporting within 30 days after the person becomes aware of the event.
- Alternatives to product recalls include a safety alert, product notification, product withdrawal, product recovery and user information sessions/materials.
If a recall is issued, the company’s Authorised Representative (AR) will be responsible for all recall-related activities including communication with SAHPRA, hence all licence holders have to ensure that AR information is always up to date. No recall actions can be completed without SAHPRA’s acknowledgement and involvement as there are different classifications and types of recalls that warrant different actions.
In cases where multiple distributors/wholesalers sell the same product, ARs from the various license holders need to be involved and are required to collaborate. This would involve a reconciliation of records from the various companies to ensure appropriate actions and follow through.
Products on a licence which were recalled prior to the local distributor importing the product would also need to be reported to the regulator. SAHPRA works with international bodies to stay abreast of international recalls. The local licence holder would need to maintain records of the international recall as these may need to be used for future decisions on changes required before the product may re-enter the market.
Wholesalers could report to SAHPRA directly, however, the distributor or manufacturer would need to be involved to ensure alignment on actions and responses across the product supply chain.
There was no set format for the submission of a report but referring to this guideline will ensure that all the required information is submitted. Internal quality management forms or a company letterhead are acceptable to use when sending the information to SAHPRA.
Attendees questioned if SAHPRA could make use of an online system for the management of adverse event reporting. Ms Julsing Keyter agreed that this would be valuable, although due to capacity restraints SAHPRA’s priority was to implement an online license application process.
Key persons to whom to report adverse events are:
- Mlungisi Wondo, Recall: Mlungisi.Wondo@Sahpra.org.za
- Momeena Omarjee, Acting Deputy Director: Medical Devices: Momeena.Omarjee@Sahpra.org.za
- Andrea Keyter, Senior Manager: Medical Devices & Radiation Control: Andrea.Julsing@Sahpra.org.za
When questioned on the actions to take should SAHPRA not acknowledge a recall from a licence holder, Ms Julsing Keyter explained the escalation process to be followed: first to Portia Nkambule (Chief Regulatory Officer) and then Dr Boitumelo Semete-Makokotlela (SAHPRA CEO). It was pointed out that SAHPRA may not respond if the member’s communique was merely a report or notice, but that SAHPRA must respond if the case warrants a recall, regardless of the regulator’s capacity restraints.
Ms Simone Rudolph Shortt, a regulatory consultant with ISO Health SA, reiterated the regulatory requirements presented by Ms Julsing Keyter and provided practical implications for the actions that needed to be taken after sending a report to SAHPRA.
Vigilance or post-market surveillance on product incidents and understanding the different definitions of an incident in relation to clinical performance and performance evaluation would guide the license holder’s Authorised Representative on action to take. Ms Rudolph Shortt linked the role of the AR to that covered in a previous SAMED-SALDA Regulatory Forum and the responsibilities of that AR in the vigilance and recall process. “Proper investigation of an incident is essential as this helps determine if an incident is reportable or not.” This would also determine if preventative and/or corrective actions would need to be taken.
Adverse events were incidents outside of the norm, that were not expected to happen, or the instructions for use omit that the product could lead to death, permanent impairment or serious injury. A near incident is one that might have led to death or serious injury but was avoided due to the healthcare practitioner’s intervention. In this case, further testing and examination will need to be done to establish the full extent on why the near incident occurred. Based on internal investigations, the AR can determine if a report needs to be made to SAHPRA, consider various exemptions and propose preventative and corrective actions which could include product recalls.
Mr Dirk Gey van Pittius, Senior Regulatory Manager for Medtronic SA, provided an overview of the international landscape when it came to vigilance and recall. Major players in this arena included the United States Food and Drug Administration (FDA), Health Canada, the European Union (EU) and the International Medical Device Regulators Forum (IMDRF).
In his introduction, Mr Gey van Pittius said it is important to note that the manufacturer’s regulatory obligations continue throughout the device’s lifecycle and long after the sale to a customer. Activities, definitions and requirements on post-market activities are not always internationally harmonised and persons responsible need to keep abreast of individual considerations.
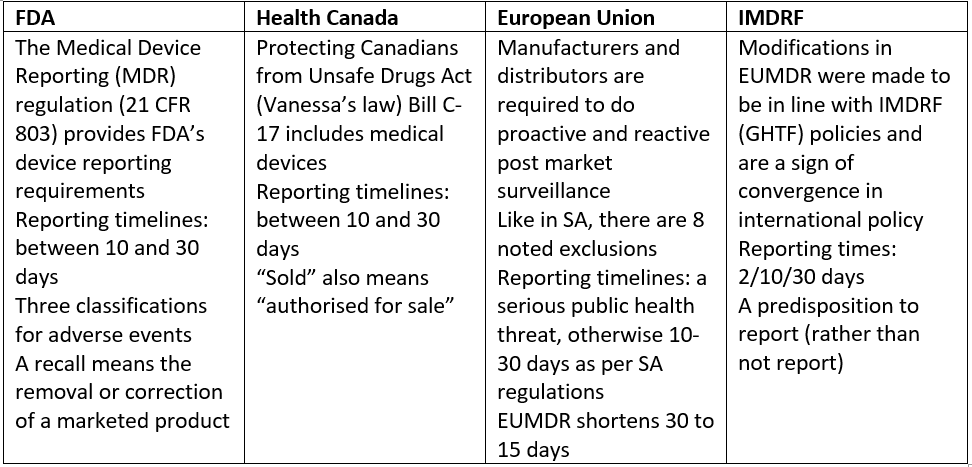
As showcased above, the processes of vigilance and recall are not unique to South Africa and there is significant overlap in global practice. An effective post-market surveillance system is important for better health and safety of patients and users, to reduce the recurrence of incidents and support appropriate corrective and preventative actions.
In closing, Mr Gey van Pittius noted: “Post-market lifecycle management may be the most resource-intensive phase for medical device manufacturers and distributors.”